Methods to Analyze Multi-Omicis Data
Multi-Omics
Multi-Omics指同时获得两个或两个以上组学数据,如基因组、转录组、蛋白质组等,并将它们结合在一起分析、挖掘,以获得更全面、更系统的生物学解释、分子作用机制等。
多组学研究的意义
整合多组学数据,提供不同层次的生物分子信息,有望系统地、整体地理解复杂的生物学。
多组学研究以顺序或同时的方式结合各组学数据,以了解分子之间的相互作用。
有助于评估从一个组学水平到另一个组学水平的信息流,从而有助于弥合从基因型到表型的差距。
多组学数据能够为研究提供整体视角,有助于提高疾病表型的预后和预测准确性,进而帮助更好地治疗和预防。
多组学研究的困难
多组学的研究通常与非组学数据相关联,如临床数据等,这导致多组学研究面临许多困难。
非组学数据复杂且主观定义
非组学数据通常十分复杂且主观性较强,需要标准化后将其加入预测或分类模型中。流行病学数据受制于调查模式、调查问题标准化以及背景,这可能会影响数据质量和可比性,并最终影响这些变量在结果预测中的贡献。不同研究者或组织由于收集程序不一致,也可能产生差异。临床数据受到其定义复杂性的影响,需要有统一的标准。目前有一些标准化的规则可供使用,如Clinical Data Interchange Standards Consortium (CDISC),Study Data Tabulation Model (SDTM),Analysis Data Model (ADaM)。这些非组学数据是基于评估者的技能和先前知识的复杂阐述过程产生的主观评估,这可能会导致报告偏差。
非组学数据具有异质性
非组学数据缺乏统一性,如不同尺度测量的定性或定量变量等,在分析之前需要对它们进行数据转换或归一化步骤。
非组学数据规模大
目前,许多多组学数据与非组学数据相结合的研究方法基于小尺度、低纬度的数据,而现在的非组学数据已经成为“大数据”——大规模,高维度。可穿戴监测装置和EHR (e-health records)的出现使非组学数据更易获取并向更高维度发展,且在处理客观和主观特征以及结构化和非结构化数据方面也更具有挑战性。
非组学数据的高维度也意味着存在
- 变量之间的相关结构
- 大规模纵向数据
- 数据稀疏性(即药物、实验室或诊断测试)
- 与组学数据相比,数据缺失与参与个体无关
这些因素都需要在后续分析建模时,将其考虑在内。
将组学和非组学数据联系
在病例对照设计中,组学数据和非组学数据的整合可能会受到确认偏差的影响。
组学和非组学数据之间的相互作用也会对研究产生影响。在建模的过程中,重要的是要将这种相互作用考虑在内。这些相互作用也可能非常复杂,如基因表达变化可能意味着表型异常,这导致分子数据和临床数据之间的关系更加复杂。
其它困难
每个变量或块对结果的贡献是否应该相等。
当与高通量数据集结合时,如何防止临床变量被惩罚(penalized)。
亚型的出现会给模型增加复杂性,在建模时是否要将亚型考虑在内。
多组学数据库
多组学数据覆盖基因组、蛋白组、转录组、代谢组和表观遗传组的数据,含有这些组学数据两种或两种以上的数据库可成为多组学数据库。
TCGA
The Cancer Genome Altas (TCGA) 是最大的多组学数据库之一,涵盖超过 33 种不同类型癌症的20000个个体肿瘤样本。TCGA旨在产生、整合、分析和解释肿瘤样本产生的DNA、RNA、蛋白质及表观遗传数据的特征以及临床和组织学数据。
CPTAC
Clinical Proteomic Tumor Analysis Consortium (CPTAC) 是通过对TCGA库中的生物样本通过质谱技术进行分析,获取的蛋白质组数据。如果实验中同时测得基因组数据,也可在该库获得。
ICGC
International Cancer Genomics Consortium (ICGC) 从20383名捐献者的21个原发癌位点的76个癌症项目中协调大规模生成基因组研究。
CCLE
Cancer Cell Line Encyclopedia (CCLE) 包含947种人类细胞系和36种肿瘤的基因表达、拷贝数和测序数据。还包含479种癌细胞系中24种抗癌药物的药理学特征。
METABRIC
Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) 包含来自乳腺肿瘤的临床特征、表达、单核苷酸多态性和拷贝数变异数据。
TARGET
TARGET 包含 24 种癌症分子类型的临床信息、基因表达、miRNA 表达、拷贝数和测序数据。
OmicsDI
Omics Discovery Index (OmicsDI) 包含来自公共数据结构中的 11 个存储库的数据集。是一个开源平台,用于访问、发现和整合基因组学、转录组学、蛋白质组学和代谢组学数据集。包含来自人类、模式生物和非模式生物的数据集。
多组学数据分析方法
根据算法思想,可将目前已有的多组学数据分析方法分为几个大类: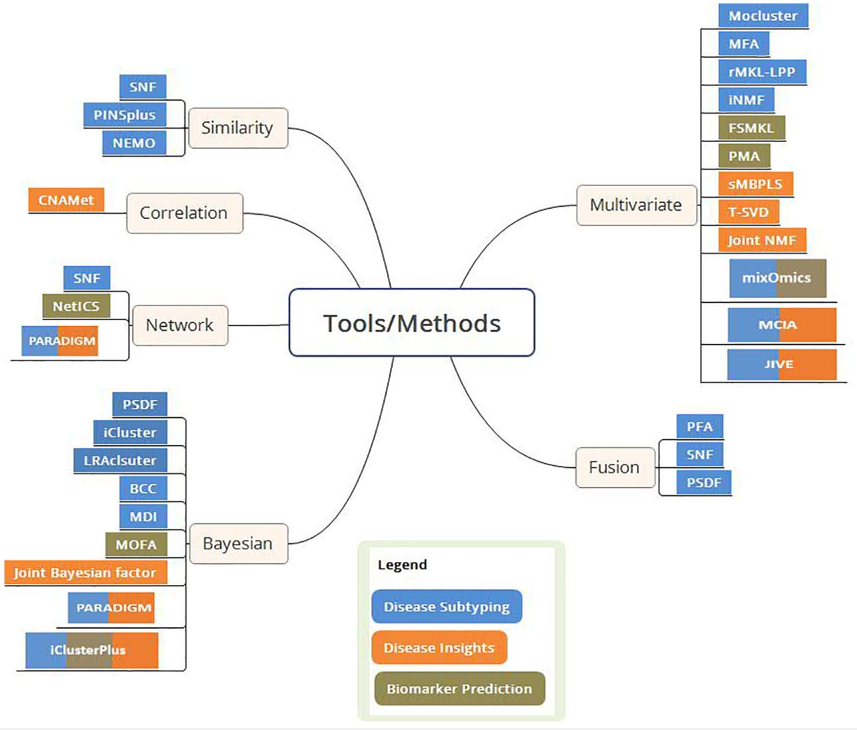
Network Based
SNF (Similarity network fusion)是一种基于网络的方法,使用网络融合方法整合多组学数据集。它为每种数据类型创建一个单独的网络,然后使用非线性网络融合方法将它们融合成一个单一的相似性网络。 融合步骤基于消息传递理论,使网络在每次迭代中更像其他网络。
NetICS (Network-based integration of multi-omics data)为基于网络的多组学数据集成提供了一个框架,用于癌症基因优先排序。可预测遗传畸变、表观遗传变化和miRNA对相互作用网络中下游基因和蛋白质(表达)的影响。在有向功能交互网络上使用每个样本的网络扩散模型,并通过聚合个体排名得出种群水平的基因排名,并为所有样本提供全局排名。
Bayesian approach
iCluster根据多种数据类型同时推断,为样本生成一个单一的聚类分配。这种无监督方法使用联合潜在变量模型进行集成聚类,并在单个框架中结合不同数据类型之间的关联以及数据类型内的方差-协方差结构,同时降低数据集的维度。通过期望最大化算法获得似然推理。
iClusterPlus是iCluster的增强版,使用广义线性回归来确定综合基因组、表观基因组和转录组分析的分类和数字(连续和计数)变量的联合模型。该方法使用一组潜在变量”k”来代表驱动因素,这些因素预测关键的基因组变量,从而捕捉生物变异。此外,使用Lasso回归方法,iClusterPlus确定了有助于亚型之间生物变化的特征子集。
LRAcluster使用概率模型与低秩近似法来寻找主要的低维子空间,以对全基因组数据进行分类。在这种方法中,每个组学数据都以大小匹配的参数矩阵为条件,并且这个低秩参数矩阵可以在低维空间中表示。 用户定义的维度参数(基于数据的解释方差)和聚类数(基于轮廓值)有助于更快地降维和更好地聚类疾病亚型。
PSDF (Patient-specific data fusion)该方法使用贝叶斯非参数模型(Dirichlet 过程混合模型)来整合CNV和基因表达数据,以将样本分层为子组。每个样本根据它们在2个数据集之间的一致性被分配一个二元状态。只有表现出一致性的样本融合在一起,而其他样本保持未融合,因此考虑了患者特定的融合模型。
BCC (Bayesian consensus clustering)提出了一种数据驱动的共识聚类方法,该方法对源特定特征以及使用有限狄利克雷混合模型扩展以解释多个数据源的整体聚类进行建模。
Joint Bayesian factor使用非参数贝叶斯因子分析来整合组学数据集。 这种方法使用 beta-Bernoulli 过程将特征空间分解为共享的和特定于数据的组件。
MDI (Multiple dataset integration)使用 Dirichlet 混合模型对每个数据源进行聚类,同时对聚类之间的成对依赖性进行建模。MDI在分配给组件(如基因组特征)的变量级别链接模型。组件变量级链接允许捕获多组学数据之间的依赖关系。
MOFA (Multi-omics factor analysis)是一种无监督方法,用于在相同或部分重叠的样本上整合多组学数据类型。
PARADIGM的应用可以扩展到对所研究疾病的发现。
Fusion-based approaches
PFA (Pattern fusion analysis)允许在低维特征空间中跨异质基因组谱识别集成样本模式。首先使用主成分分析获得局部样本模式。 其后将这些局部样本模式与公共特征空间对齐,并跨大多数数据类型合成全局样本模式。在此过程中,将定量测量每种数据类型(或单个样本)对全局样本频谱的贡献,并迭代降低偏差或系统噪声的影响以更好地拟合数据。
Similarity-based approaches
PINSPlus (Perturbation clustering for data integration and disease subtyping)是一种无监督的聚类方法,有助于从多组学数据中识别亚型。为了识别亚型,该算法确定患者在单个集群中分组的频率(1)当数据受到干扰时,(2)使用不同类型的组学数据时,(3)使用不同的聚类技术时。 所有场景中的强关联患者都聚集在一起形成一个亚型。
NEMO(Neighborhood-based multi-omics clustering) 是一种基于相似性的简单多组学聚类方法,它进一步建立在先前建立的聚类方法(如 SNF 和 rMKL-LPP)的基础上。该方法最初为每个输入组学数据集构建基于患者间相似性矩阵的欧几里德距离。然后将每个组学的相似性矩阵整合到一个矩阵中,然后使用光谱聚类方法对其进行聚类。
Correlation based approaches
CNAmet用于对拷贝数改变、DNA 甲基化和基因表达数据进行综合分析。
Other multivariate approaches
mixOmics提供了一组有监督和无监督的多元方法来执行多组学数据集的整合,重点是变量选择。
moCluster使用多表多元分析方法来识别跨多组学数据集的模式。
MCIA (Multiple co-inertia analysis)是一种探索性数据分析方法,它捕捉多个高维数据集(如基因表达、miRNA表达、蛋白质表达)之间的相互关系。
JIVE (Joint and individual variation explained)通过分离数据集的联合效应和个体效应来整合多组学数据。
MFA (Multiple factor analysis)是一种通过将其投影到低维变量空间来帮助整合组学数据集的方法。
rMKL-LPP 使用多核学习来集成异构多数据并执行子类型识别。
iNMF (Integrative nonnegative matrix factorization)扩展了NMF框架以在集成多个数据时考虑异构效应。
FSMKL (Feature selection multiple kernel learning)是一种监督分类方法,使用多个内核来捕获数据集之间的相似性,以识别疾病进展的特征。
sMBPLS (Sparse multi-block partial least squares)允许多块输入包含多个调控组学数据集,例如 CNV、DNA 甲基化和调控基因表达的 miRNA 表达。
T-SVD (Thresholding singular value decomposition)有助于识别 2 个组学数据集之间的调控机制,尤其是当调控特征大于测量样本时。
Joint NMF 该分解框架从多个数据集(相同样本)中识别相关模块,以推导出 md 模块,以揭示潜在的多层监管因素。
参考文献
[1] Subramanian I, Verma S, Kumar S, Jere A, Anamika K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. January 2020. doi:10.1177/1177932219899051
[2] López de Maturana E, Alonso L, Alarcón P, Martín-Antoniano IA, Pineda S, Piorno L, Calle ML, Malats N. Challenges in the Integration of Omics and Non-Omics Data. Genes (Basel). 2019 Mar 20;10(3):238. doi:10.3390/genes10030238