Batch Effects Matter and Avoid Them in Omics Data
什么是批次效应
批次效应是指实验子组在不同的实验条件之下具有与研究中的生物学或其它科学变量无关的性质不同的行为,即实验中测量值之间由于技术因素造成的系统性差异。
什么情况下会有批次效应
批次效应在生物学实验中广泛存在,无论是microarray expression profiling还是mass spectrometry产生的数据,都观察到明显的批次效应。具体到研究疾病的差异基因/蛋白或变异数据(如拷贝数变异)的研究中同样观察到明显批次效应。
造成批次效应的因素有很多,试剂批次不同、实验时间不同、仪器状态变化、实验员不同都可能造成批次效应。其中一些批次效应能够通过规范化实验操作、更好的实验设计避免;另一些则需要通过对所得数据进行处理才能够消除。
在Leek等人对已公开数据批次效应的研究发现,已公开的数据中存在明显批次效应。且在许多实验条件和技术中,技术性因素比生物性因素对实验结果更具影响力。当批次效应发生时,常常与生物性因素混淆,导致下游研究结果不准确。
为什么要关注批次效应
当批次效应发生时,可能会
- 增加变化(variability)而掩盖真正生物学信号,导致得到错误的生物学或临床结论
- 与特征信号混在一起,导致下游分类器构建困难
- 阻碍生物学上重要亚型的发现或与亚型混淆难以区分
- 导致实验资源分配不当,结果缺乏可重复性
因此,消除批次效应对得到准确、可重复性高的结论非常重要。
如何避免或处理批次效应
实验设计
避免出现批次效应的首要步骤是合理的实验设计。高通量的实验应在实验设计时考虑到批次效应,在实验过程中尽可能避免批次效应的出现。实验分组时也需保证平衡性(balance),避免非研究目标的生物学因素对实验结果造成影响,进而和批次效应混杂,导致数据难以处理。
数据处理
优秀的实验设计是降低批次效应的基础,在此基础之上处理批次效应主要有以下两个步骤:
- 识别并量化数据中潜在的批次效应,包括人为因素
- 使用已知或找到的人为因素调整数据以适应下游分析
识别并量化批次效应
识别和量化批次效应主要通过主成分分析(Principal Component Analysis, PCA)或其它数据可视化工具如聚类、多维数据标化等。
如果数据处理结果中出现:
- 样本按处理组或时间聚类
- 大量特征与处理组或时间高度相关
- 主成分与批次处理组或时间相关联
则表明数据中极大可能存在批次效应,必须在下游数据处理前考虑到批次效应的影响。
处理批次效应
根据其算法思想可将消除批次效应的方法分为如下几个大类:
- 简单线性模型(Simple Linear Models): Mean-scaling, zero-centering
- 经验贝叶斯方法(Empirical Bayes): ComBat
- 因子分析(Factor-based analysis): Surrogate Variable Analysis (SVA), Removed unwanted variation (RUV)
- 深度学习(Deep Learning): NormAE
其中,前两种消除批次效应的方法需要已知造成批次效应的因素,如实验时间等。
根据先验知识、实验数据规模、特征空间大小、研究目的等因素,可挑选不同的处理批次效应的方法:
- 大数据;特征空间有限;有限的生物异质性;批次效应或分类影响因子已知;目标是简单分析:Two-way ANOVA
- 小数据;特征空间有限;有限的生物异质性;批次效应或类影响因子已知;目标是去除批次效应:ComBat
- 中/大型数据集;大特征空间;存在生物异质性;类因子已知,批次效应影响因素不必已知;目标是移除批次效应且确定批次效应影响因素:SVA, RUV
- 中/大型数据集;大特征空间;存在生物异质性;批次效应或分类影响因子都不必已知;目标是移除批次效应且确定批次效应影响因素但不需直到类因子:unsupervised methods (PCA), RUV
算法补充: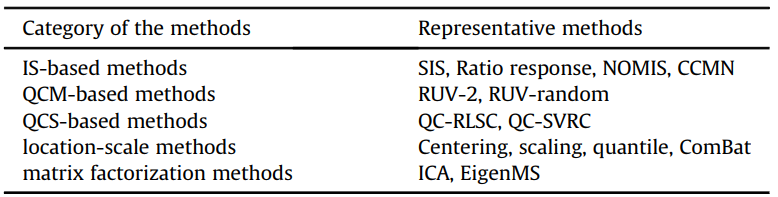
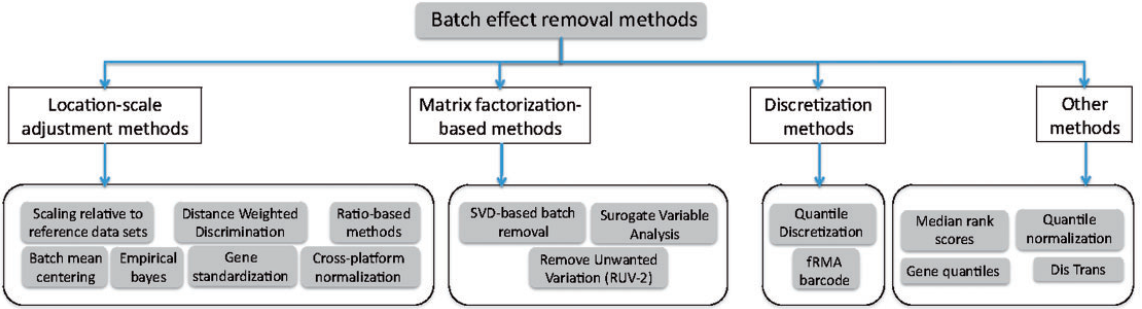
批次效应处理评估
消除批次效应之后,通常会检查处理的效果。常用的方法有PCA、层次聚类等,检查数据是否有与批次相关联的偏移、聚类结果是否更符合生物学先验知识等。
评估方法小结: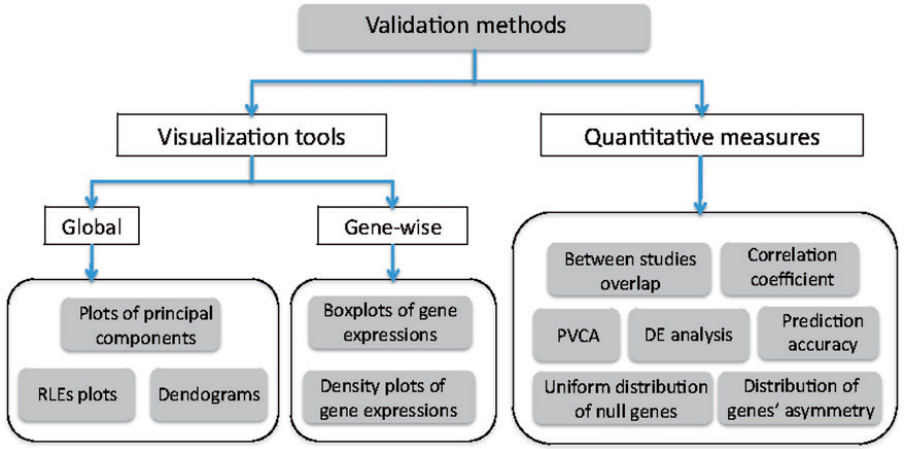
批次效应处理流程
Jelena Čuklina等人针对批次效应的研究中提供了一个消除蛋白质数据批次效应的处理流程和一个包含所有处理步骤的R包——proBatch (Bioconductor, Docker container, GitHub repository都可获取该R包)
流程图总览
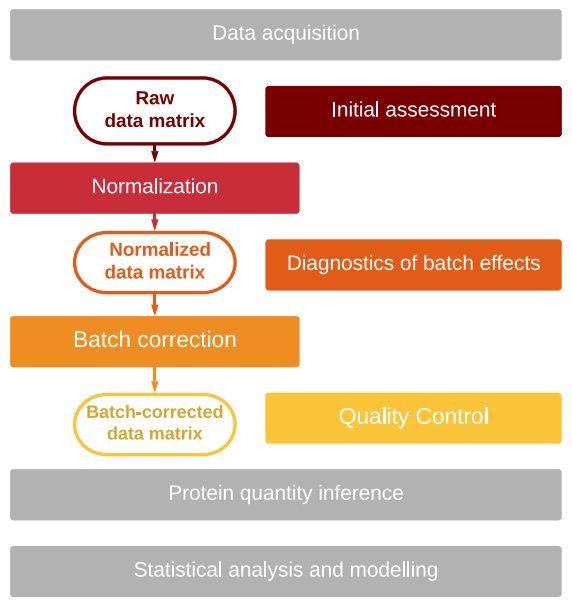
- 初步评估原始数据中是否存在批次效应
- 归一化使数据集中所有数据在同一尺度
- 归一化后数据评估,以确定数据是否需要进一步处理
- 批次效应校正以纠正特征偏移
- 质量控制测试:是否在保留有意义信号的同时减少了数据偏差
Raw Data Matrix
在进行这一流程之前,应当先对数据进行预处理,如肽段识别、肽段定量、FDR筛选、log-transformed或variance stabilizing transformation。
虽然在ion fragment、peptides、protein层面都可以识别并处理批次效应,但因为这个过程改变了对蛋白质推断至关重要的特征丰度,最好针对ion fragment或peptides数据处理批次效应。且在处理过程中应包含尽可能多的数据以保证数据分布最接近真实情况。
Initial Assessment
这一步骤主要目的是确定数据的偏移程度并确定一个归一化方法。通常情况下,样本间intensity会有一定差异,调整这种差异有助于数据的比较,能够更好地识别出需要进一步处理的因素。主要方法有三种:
- 按照质谱测量或技术批次的顺序绘制样品强度平均值或中值,评估每个批次中的质谱漂移或离散偏差
- 箱线图,评估样本方差和异常值
- 批次间与批次内样本相关性
通过上述步骤来检验
- 数据分布是否具有一致性
- 样本间的相关性
- 如果有差异,这种差异是否与批次相关联
Normalization
归一化的目的就是将所有样本的数据处于同一尺度,使得跨样本数据能够互相比较。常用的归一化方法有quantile normalization、median normalization和z-transformation,应根据数据的异质性和数据分布情况选择合适的归一化方法。
数据的异质性
- 数据相似程度高: quantile normalization
- 数据具有本质差异: HMM-assisted normalization
- 数据会有包含信息的异常值(outliers): 选择能够保留异常值与总体数据关系的归一化方法
样本丰度分布
- 通常情况下,只需调整数据中位数或平均值
- 如果出现方差差异较大,也需要将它们调整到同一尺度
归一化步骤应尽可能简单直接,对数据的操作越少,越能够保留数据的真实情况。归一化后可通过diagnostic plots和quality control方法对数据进行评估。
Diagnostics of normalized data
评估归一化后的数据以确定是否需要进一步的处理,主要方法为:
- Hierarchical clustering: 将相似的样本分组为树状结构,观察聚类结果是否和批次相关
- Principal Component Analysis(PCA): 观察主成分是否与批次相关联,对评估聚类依据是生物还是技术因素及检查重复组相似性十分有效
上述方法要求数据中没有缺失值,而蛋白质组数据通常含有缺失值。填补缺失值时应特别注意尽可能保留数据原有的分布,不能盲目填0或一个较小的随机数。
蛋白质组通常使用肽段数据检查是否存在与批次相关的偏移。如在DIA数据中加入iRT肽段用于数据特征校正。由于不同肽段对不同批次效应的response不同,有必要检查大量肽段以确定是否存在批次效应。通过检查肽段数据还能够确认跑样顺序是否对数据造成影响,是否有与顺序相关的变化趋势(trends)。
还可以用ion fragments的数据校正蛋白数据,但目前针对肽段的方法更加广泛。
Batch effect correction
归一化能够校准数据的全局,而批次效应校正主要针对特征峰和特征组。根据批次效应的形式,可将消除批次效应的方法分为两大类:
- Continuous
消除连续的批次效应主要通过拟合的方式,如LOESS fit,或使用其它连续算法。
在质谱大数据(hundreds of samples)中会出现信号漂移现象,这仍是一个亟待解决的问题。 - Discrete
消除离散的批次效应时常用mean and median centering。
基于贝叶斯模型的ComBat算法也能够用于处理蛋白质数据,但需要已知所有的批次效应影响因素。
Quality control
质控步骤主要用于评估归一化和批次效应校正之后的数据质量,好的数据校正应做到消除偏移(negative control)和提升数据(positive control)两方面。
- 消除偏移的标准
- 聚类或PCA后,同组数据聚集依据与批次无关,更多地受生物学因素影响
- 肽段(或其他特征,如ion fragments)没有与批次相关的偏移
- 提升数据
- 通常情况下,数据提升的标准为聚类结果更符合生物学先验知识,后续差异检验步骤能够识别出更多差异。但这种标准并不具有足够的客观性,尤其是后一个标准并不一定表明数据提升,还有可能是false positive
- 交叉验证:差异表达蛋白或最佳分类特征蛋白列表高度重合。但这种方法依赖于数据集和特征空间的大小,当数据集较小,后一个蛋白列表本身不稳定,会对评估造成影响。在另一篇综述中强调应避免交叉验证来评估数据质量
- 检查重复组间的方差:如果数据的归一化和消除批次效应步骤合理,重复组间的方差应降低
- 样本间相关性:技术或生物学重复样本间的相关性应明显高于与其它样本的相关性
- 距离矩阵:与上一方法的逻辑相似,但通过计算样本间距离来评估数据
- 肽段间的相关性:来自同一蛋白的肽段具有正相关或高相关性,而来自不同蛋白或不相关的肽段间的相关性应接近于0
消除批次效应的方法/工具
ComBat
简介
ComBat基于参数和非参数经验贝叶斯框架,用于调整具有批次效应的数据。该方法对小样本中的异常值具有健壮性,并且与大样本的现有方法相当。
以Microarray为研究对象。
经验贝叶斯框架使用基础
- 已经广泛用于大规模microarray数据:稳定具有极高或极低比率的基因的表达比,缩小所有其他基因的方差来稳定基因方差避免伪影的影响等
- 已有批次效应处理方法(如SVD、DWD和L/S method)需要大批量数据,且不能兼容小批次数据的离群值
- 对高位效数据组有较强的健壮性
- 利用跨基因和跨样本的“借用信息”,以得到更好的估计或更稳定的结果
基本假设
- 结合基因间常见的系统批次效应,假设导致批次效应的现象通常以相似的方式影响许多基因
- 通过汇集信息(pooling information)来估计代表批次效应的L/S模型参数,在每个批次的基因中缩减(shrink)批次效应参数估计值,使之朝着批次效应估计值的总体平均值(跨基因)发展
- 数据已被归一化,所有样本的基因表达值已被估计
关键步骤
- Standardize the data
- EB batch effect parameter estimates using parametric empircal priors
- Adjust the data for batch effects
QC-RLSC
简介
QC-RLSC(quality control-based robust LOESS signal correction)可用于信号校正和整合来自不同实验批次的数据。
以Metabolics数据为研究对象。
基本原理
LOESS曲线拟合结合了经典的线性最小二乘法回归的简单性和非线性回归的灵活性。它通过对数据的局部子集进行简单的模型拟合来建立一个函数,逐点地描述数据中的确定性变化部分。不需要指定任何形式的全局函数来拟合数据的模型,而只需要拟合数据的片段。
对数据的每个子集进行拟合的局部多项式被限制为一阶或二阶(即局部线性或局部二阶),并使用加权最小二乘法拟合(标准三立方权重函数)。
优化平滑参数(或称跨度)以获得更好的回归曲线。不使用过小的平滑参数以避免曲线受到随机误差的影响。
留一法交叉验证(leave-one-out cross validation)以避免过拟合。
关键步骤
- 在实验结束、色谱解卷积后,使用QC-RLSC对数据进行标准化(normalize)
- 依据注入顺序,对质控数据进行LOESS拟合
- 对整个分析运行的校正曲线进行内插,并对该特征的总数据集进行标准化
通过这些步骤,在一次分析中,峰响应的任何衰减都被最小化。
ICA
简介
使用时空独立成分分析(spatio-temporal independent component analysis)对批次效应建模,并移除这些影响。
以microarray datasets为研究对象。
基本原理
对整合的数据集进行因子分解(factorization),移除与子数据集具有某种相关性的组件,以获得最终数据集。这一过程能够从数据中移除批次效应。
去除的成分是可以解释的,很容易检查它们是否与某些感兴趣的生物信息相关。
ICA被证明能更好地模拟不同的变异(variables)来源。
关键步骤
假设汇总的数据集是一个按样本划分的基因矩阵X,Xi,j 表明gene i 在样本 j 中的表达量。
WaveICA
简介
WaveICA基于带有小波变换的独立成分分析,作为大规模代谢组学数据的阈值处理方法捕获并去除批次效应。
研究对象为Metabolics数据。
基本原理
利用样品在注射顺序中的时间趋势,将原始数据分解为具有不同特征的多尺度数据,提取并去除多尺度数据中的批次效应信息,获得干净的数据。
在实际问题中DWT有局限性,WaveICA中使用的小波变换为Maximal overlap discrete wavelet transform (MODWT)。
关键步骤

NormAE
简介
Normlization Autoencoder(NormAE)基于非线性自编码器和对抗性学习的新型深度学习模型。
研究对象为Metabolics数据。
基本原理
将非线性自编码器和DNN结合,提高模型的非线性拟合能力,使得批次效应结果和其它因素影响结果分开,以在去除批次效应的同时保留更多生物学特征。
在AE模型的训练过程中,训练一个额外的分类器和排序器来对抗性正则化,潜在的特征被编码器提取出来,然后解码器在没有批次效应的情况下重建数据。
关键步骤
黑色实线和红色虚线分别表示反向传播算法的前向和后向计算步骤。蓝色虚线路径表示训练后的批量效应去除步骤。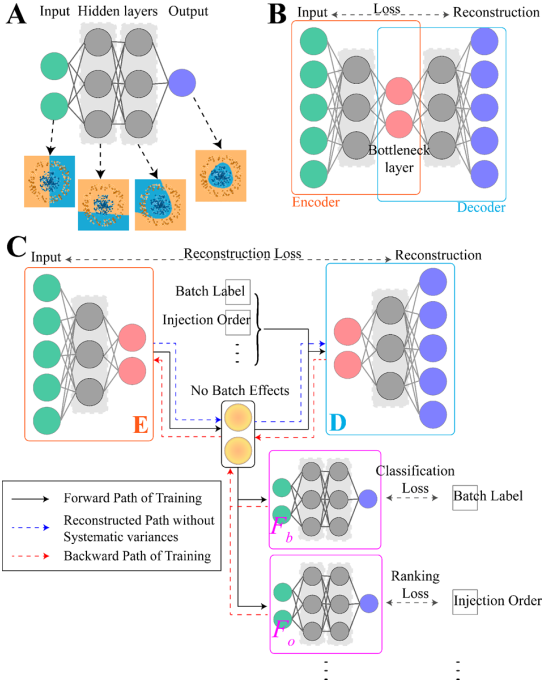
参考文献
[1] Leek, Jeffrey T et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nature reviews. Genetics, vol. 11,10 (2010): 733-9.
[2] Čuklina, Jelena et al. Diagnostics and correction of batch effects in large-scale proteomic studies: a tutorial. Molecular systems biology, vol. 17,8 (2021): e10240.
[3] Zhou, Longjian et al. Examining the practical limits of batch effect-correction algorithms: When should you care about batch effects?, Journal of Genetics and Genomics, Vol. 46, 9(2019): 433-443.
[4] Goh, Wilson Wen Bin et al. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol, Vol. 35, 6 (2017):498-507.
[5] Johnson, WE et al. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, Vol. 8, 1 (2007): 118–127.
[6] Dunn, W et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocol, 6 (2011): 1060–1083.
[7] Sompairac, Nicolas et al. Independent Component Analysis for Unraveling the Complexity of Cancer Omics Datasets. International journal of molecular sciences. vol. 20, 18 (2019): 4414.
[8] Deng, Kui et al. WaveICA: A novel algorithm to remove batch effects for large-scale untargeted metabolomics data based on wavelet analysis. Analytica Chimica Acta, Vol. 1061, (2019): 60-69.
[9] Rong, Zhiwei et al. NormAE: Deep Adversarial Learning Model to Remove Batch Effects in Liquid Chromatography Mass Spectrometry-Based Metabolomics Data. Analytical Chemistry, Vol. 92, 7 (2020): 5082–5090.